

Авторы: О.И. Виноградская1, В.В. Фадеев1, Д.В. Липатов2
Authors: O.I. Vinogradskaya1, V.V. Fadeyev1, D.V. Lipatov2
Организации:
- Первый МГМУ им. И.М Сеченова
- ФГБУ ЭНЦ
Author affiliation:
- First MSMU I.M. Sechenov
- Endocrinology Research Centre
Abstract:
The pathological processes in Graves’ orbitopathy (GO) include inflammatory infiltration of retro-ocular tissues within the orbit. Glucocorticoids (GCs) are the most common immunosuppressants used in the treatment of active and severe GO. They are most effective when given early in the course of the disease. Although intravenous (i.v.) schedule is much better tolerated than the oral one, the numerous side effects of i.v. steroid use have been reported. Glucocorticoid-induced hyperglycemia is a common adverse effect. Recent demonstrations that even shortterm elevations in blood glucose level may be associated with adverse sequelae. GCs are well known to reduce insulin sensitivity, resulting in increased hepatic glucose production and decreased peripheral glucose disposal. The role of β-cell dysfunction in GC-related diabetogenic effects is less clear. The present paper offers an overview of the possible mechanisms of GC-induced hyperglycemia. Based on the study performed in our clinic the relation between i.v. steroids in GO and hyperglycemia is discussed.
Эндокринная орбитопатия (ЭОП) представляет собой аутоиммунное воспаление ретробульбарных структур: жировой клетчатки и глазодвигательных мышц. В большинстве случаев заболевание имеет легкое течение и не требует активного лечения. При активной ЭОП средней тяжести применяется иммунносупрессивная терапия, в первую очередь глюкокортикоидами (ГК).1 Патофизиологическим обоснованием применения ГК при ЭОП является их способность активно взаимодействовать с системой иммунитета и подавлять воспалительные реакции. Из всех противовоспалительных препаратов только ГК действуют на большинство известных цитокинов — блокируют активность «провоспалительных» цитокинов и повышают активность «противовоспалительных».
Препаратом выбора при ЭОП является 6-метилпреднизолон, обладающий минимальной минералкортикоидной и мощной противовоспалительной активностью и, в отличие от других ГК, сбалансированными геномными и негеномными эффектами.2 Данные эффекты реализуются в зависимости от дозы. Так, в низких концентрациях (>10-12 моль/л) ГК реализуют свое действие за счет геномных эффектов, для развития которых требуется более 30 минут. Считается, что геномный механизм осуществляется посредством связывания ГК со специфическими цитоплазматическими рецепторами, после чего комплекс ГК-рецептор проникает в ядро клетки, где взаимодействует с участками ДНК, расположенными в промоторном фрагменте стероид-отвечающего гена (glucocorticoid response element), и регулирует процесс транскрипции определенных генов, оказывая влияние на синтез белков, в том числе ферментов: снижается выработка провоспалительных ферментов- коллагеназы, эластазы, активатора плазминогена. Одним из наиболее важных белков, повышенный синтез которого опосредуется через иРНК, является липокортин, ингибирующий синтез простагландинов, лейкотриенов, тромбоксана и кислородных радикалов. 2 В высоких концентрациях (>10-4 моль/л), наряду с геномным эффектом, ГК обладают негеномным эффектом. Негеномные эффекты являются результатом прямого физико-химического взаимодействия ГК с биологическими мембранами или стероидселективными мембранными рецепторами. К негеномным эффектам относится стабилизация клеточных мембран и мембран органелл, снижение проницаемости капиллярного эндотелия, подавление миграции лейкоцитов в очаг воспаления за счет угнетения экспрессии молекул адгезии, снижения функциональной активности эндотелиоцитов, моноцитов/макрофагов, нейтрофильных гранулоцитов и фибробластов. 2
При этом удельный вес геномных и негеномных эффектов в механизме действия синтетических ГК неодинаков (рисунок 1). При применении ГК в дозах выше 250 мг (пульс-терапия) негеномные эффекты играют весьма значительную роль в получении быстрого и выраженного терапевтического эффекта. При этом сила негеномных эффектов метилпреднизолона более чем в 3 раза превышает таковую преднизолона, что подтверждает необходимость использования для пульс-терапии именно метилпреднизолона.3 Дексаметазон также обладает выраженным негеномным действием, однако его использование сдерживается неблагоприятным побочным профилем.
Рисунок 1. Относительная сила действия глюкокортикоидов

Однако одной из проблем применения ГК являются побочные эффекты, которые могут возникать даже в отдаленные сроки. Возможно развитие нарушения толерантности к глюкозе, диабета, увеличение массы тела, гастрита, бессонницы, депрессии, артериальной гипертензии, появление признаков кушингоидизации, остеопороз, дислипидемия и ССЗ, нарушения со стороны психики.4,5
В данной статье мы попытаемся разобраться в механизмах развития гипергликемии при приеме ГК, обсудить влияние пульс-терапии метилпреднизолоном при ЭОП на углеводный обмен. В своей практике мы часто сталкиваемся с тем, что из-за риска развития гипергликемии врачи откладывают или отказываются от назначения пульс-терапии при ЭОП у пациентов с исходно нормальным углеводным обменом, не говоря уже о пациентах с сахарным диабетом. В большинстве случаев этот риск довольно сильно преувеличен. С другой стороны, некоторые врачи недооценивают негативных последствий глюкокортикоид-индуцированной гипергликемии и не проводят соответствующих мероприятий по ее коррекции.
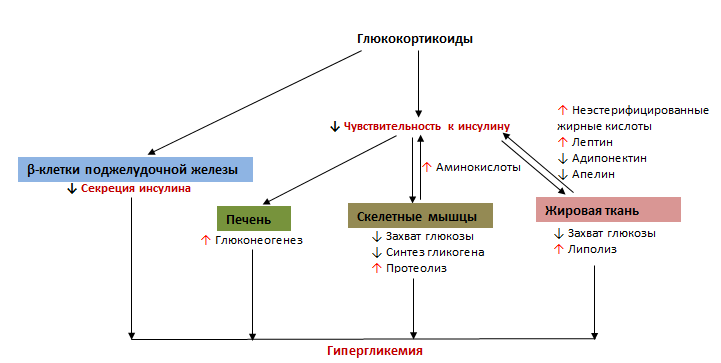
Рассмотрим механизм развития гипергликемии при гиперкортицизме. Сперва, рассмотрим влияние ГК на функцию β –клеток. Известно, что на β-клетках поджелудочной железы экспрессируются рецепторы к ГК, посредством которых модулируется экспрессия различных генов, что сопровождается снижением синтеза инсулина.6 Снижение синтеза инсулина происходит в результате:
- снижения образования GLUT-2 и глюкокиназы в β-клетках, что ведет к снижению захвата глюкозы и фосфорилирования, снижению синтеза АТФ и как следствие, к снижению притока калия внутрь β-клетки. Низкая концентрация внутриклеточного калия уменьшает деполяризацию мембраны и замедляет приток кальция,6 необходимый для экзоцитоза гранул, содержащих инсулин.7
- усиления экспрессии α2— адренэргических рецепторов, приводящей к уменьшению цАМФ, снижению активности протеинкиназы А с последующим подавлением высвобождения инсулина.
- уменьшения массы β-клеток в результате ГК-индуцированного апоптоза.8
- ГК-индуцированного липолиза, сопровождающегося повышением уровня циркулирующих триглицеридов и неэстерифицированных жирных кислот, которые накапливаясь в β-клетках приводят к нарушению их функции (липотоксичность).9
Помимо влияния на β-клетки, ГК оказывают воздействие и на периферические ткани, что сопровождается снижением захвата и утилизации глюкозы6 в результате:
- развития пост-рецепторного дефекта из-за снижения ключевых медиаторов активности инсулина в периферических тканях: субстрата рецептора инсулина 1 типа, фосфатидилинозитол-3-киназы и протеинкиназы В,10-12 что приводит к нарушению транслокации переносчиков глюкозы к поверхности клетки и снижению утилизации глюкозы.13
- ГК-индуцированного протеолиза с повышением уровня циркулирующих аминокислот,14 ингибирующих фосфорилирование субстрата рецептора инсулина и активацию фосфатидилинозитол-з-киназы на уровне гепатоцитов и миоцитов скелетных мышц, что дополнительно снижает захват глюкозы и синтез гликогена,15
- снижения фосфорилирования инсулин-стимулированной гликогенсинтазы-киназы-3 в мышечной ткани, что снижает синтез гликогена,16
- уменьшения инсулиноподобного эффекта глюкагоноподобного пептида-1,6
- повышения секреции глюкагона,6
- ГК-индуцированного липолиза с образованием большого количества циркулирующих свободных жирных кислот. Неэстерефицированные жирные кислоты (диацилглицерол, церамид), конкурируя с глюкозой за окисление внутри миоцитов, снижают захват и утилизацию глюкозы.17 Кроме того, интрацеллюлярные липиды активируют различные сериновые киназы, такие как c-Jun N-терминальную киназу (JNK) и IkB киназу-β, которые участвуют в фосфорилировании серинового участка субстрата инсулинового рецептора 1 типа, что приводит к подавлению инсулинового сигнала.18 Таким образом ГК, не только напрямую снижают чувствительность к инсулину, но и нарушают путь передачи инсулинового сигнала за счет усиленного протеолиза и липолиза.
Помимо инсулинорезистентности, ГК увеличивают продукцию глюкозы за счет:
- ГК-индуцированной инсулинорезистентности на уровне печени, которая приводит к снижению ингибирующего влияния инсулина на продукцию глюкозы печенью, особенно после приема пищи.19,20
- усиления глюконеогенеза в результате избыточной экспрессии различных ферментов глюконеогенеза: фосфоенолпируват карбоксикиназы и глюкозо-6-фосфатазы.21-23 Ген фосфоенолпируват карбоксикиназы содержит в промотерной зоне глюкокортикоид-чувствительный участок, что позволяет рассматривать фосфоенолпируват карбоксикиназу как ключевой фермент в развитии ГК-индуцированной гипергликемии.24
- повышения уровня циркулирующих аминокислот и жирных кислот, высвобожденных в результате протеолиза и липолиза (особенно аланина и глицерола), которые являются субстратами для глюконеогенеза6 (рис.2)
Рисунок 2. Механизм развития гипергликемии при терапии глюкокортикоидами6
Таким образом, введение гидрокортизона и прием больших доз преднизолона оказывает быстрый ингибирующий эффект на β -клетки, снижая секрецию инсулина в ответ на введение глюкозы и прием пищи.25,26 Более длительная терапия ГК (2-5 дня) сопровождается повышением тощаковой и стимулированной секреции инсулина, что позволяет компенсировать развивающуюся инсулинорезистентность. Секреция инсулина и чувствительность к нему оказываются сбалансированными. Однако у лиц из группы высокого риска (с ожирением, с исходно сниженной чувствительностью к инсулину или низкой стимулированной секрецией инсулина, или им еющие родственников первой линии с сахарным диабетом 2 типа (СД2)) такой компенсации становится недостаточно, что приводит к гипергликемии.25,26 «Срыв» компенсации может произойти и у лиц, не входящих в группу риска, что указывает на еще не совсем полностью изученные процессы, происходящие при приеме ГК. 26
Подводя итоги, можно сказать, что ГК ингибируют секрецию инсулина, снижают чувствительность к инсулину. Их длительный прием приводит к гиперинсулинемии, как результат компенсаторной реакции в ответ на развитие инсулинорезистентности. Постоянный прием ГК приводит к прогрессирующему снижению функции бета-клеток у предрасположенных лиц.
Риск развития сахарного диабета у пациентов, получающих хроническую терапию ГК, выше, чем в общей популяции, примерно в 1,5-2,5 раза.27 Особенно этот риск выше у пациентов с ожирением, у пациентов старшего возраста и имеющих отягощенную наследственность по диабету, а также у пациентов, получающих высокие дозы глюкокортикоидов на протяжении длительного периода времени. Тем не менее, даже прием небольших доз ГК, но на протяжении длительного времени, сопровождается нарушением углеводного обмена. Крупное популяционное исследование показало, что риск глюкокортикоид-индуцированной гипергликемии увеличивается с увеличением дозы ГК: отношение шансов для гипергликемии составило 1,77, 3,02, 5,82 и 10,34 при приеме 1-39 мг/сут, 40-79 мг/сут, 80-119 мг/сут и ≥120 мг/сут в гидрокортизоновом эквиваленте, соответственно.28
ГК широко применяются в клинической практике. Риск развития СД при использовании ГК при заболеваниях кожи составляет 23,5%, при заболеваниях органов дыхания – 14,7%, при СКВ-12,7%.29
В своей работе мы попытались проанализировать риск развития нарушения углеводного обмена при активной ЭОП средней степени тяжести на фоне пульс-терапии метилпреднизолоном.
Материалы и методы
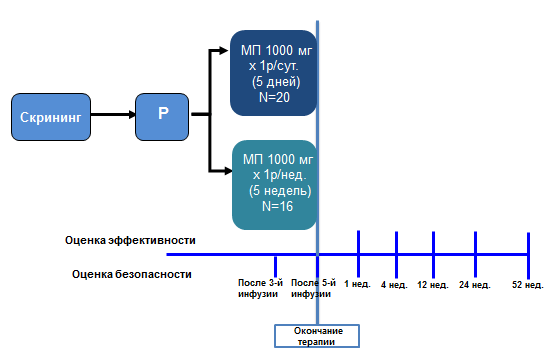
В исследование было включено 36 пациентов (27 женщин и 9 мужчин) в возрасте от 28 до 72 лет (медиана — 50 лет). Пациенты были разделены 2 группы: пациенты 1-ой группы (n=20) получали метилпреднизолон ацетат в дозе 1000 мг внутривенно капельно ежедневно в течение 5 дней. Пациенты 2-й группы (n=16) получали метилпреднизолон (МП) ацетат в дозе 1000 мг внутривенно капельно 1 раз в неделю в течение 5 недель. Суммарная доза метилпреднизолона в каждой группе составила 5000 мг. В период между инъекциями другие препараты из группы глюкокортикоидов не назначались. Пациенты обеих групп не различались по возрасту, полу, длительность ЭОП, уровню ТТГ. В каждую группу было включено по 1 пациенту с СД2 типа (р=0,69). Исходно по уровню гликемии группы между собой также не различались (таблица 1).
Таблица 1. Уровень гликемии на момент включения в исследование
| Показатель |
1 группа (МП 1 раз в день) n=20 |
2 группа (МП 1 раз в неделю) n=16 |
р |
| Глюкоза, ммоль/л | 5,0 [4,5; 5,4] | 4,9 [4,6; 5,3] | 0,66 |
Для оценки краткосрочного и долгосрочного влияния метилпреднизолона на углеводный обмен, контроль гликемии (в рамках оценки безопасности терапии) проводился на следующий день после 3-й, 5-й инфузии метилпреднизолона, через 1, 4, 12, 24 и 52 недели после окончания терапии (рисунок 3). В нашей работе оценка гликемии проводилась только натощак, постпрандиальный уровень не оценивался.
Рисунок 3. Дизайн исследования
Результаты
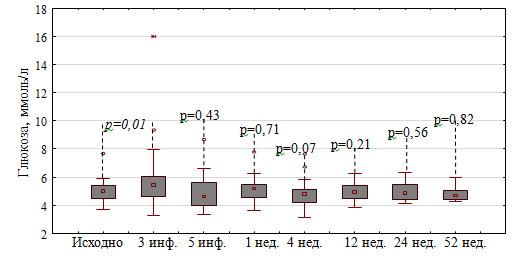
В ходе исследования при анализе уровня гликемии было зафиксировано ее повышение в первой группе после 3-й инфузии МП до 5,46 [4,58; 6,08] ммоль/л (р=0,01), максимально до 16 ммоль/л у пациента с СД2 типа, что потребовало добавление инъекций инсулина короткого действия к уже получаемой терапии метформином и глимепиридом (рисунок 4).
Рисунок 4. Динамика уровня гликемии в группе ежедневного введения МП (сравнение с исходными значениями)
Во второй группе было выявлено достоверно значимое повышение гликемии после 5-й инфузии метилпреднизолона: гликемия исходно — 4,88 [4,58; 5,28] ммоль/л, после 3-й инфузии МП — 5,08 [4,72; 5,6] ммоль/л (р=0,27); после 5-й инфузии МП — 5,41 [4,86; 5,83] ммоль/л (р=0,03) (рисунок 5).
Рисунок 5. Динамика уровня гликемии в группе еженедельного введения МП (сравнение с исходным значением)
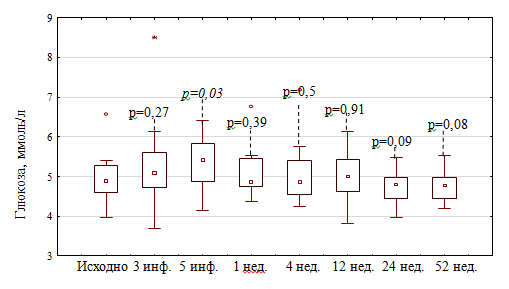
Межгрупповой разницы по уровню гликемии на протяжении всего периода наблюдения выявлено не было (рисунок 6).
Рисунок 6. Динамика уровня гликемии в обеих группах

С учетом включенных пациентов с СД2 типа, нарушение углеводного обмена (гликемия плазмы венозной крови натощак ≥ 6,1 ммоль/л) в обеих группах на протяжении всего периода наблюдения было выявлено у нескольких пациентов (рисунок 7). Относительный риск развития нарушения углеводного обмена оказался в 2,4 раза выше в первой группе, чем во второй. Однако ДИ для относительного риска составил [0,27; 20,92], р=0,39, что свидетельствует об отсутствии статистической разницы между группами по риску развития нарушения углеводного обмена.
Рисунок 7. Клинически значимое повышение уровня гликемии (≥ 6,1 ммоль/л) в обеих группах
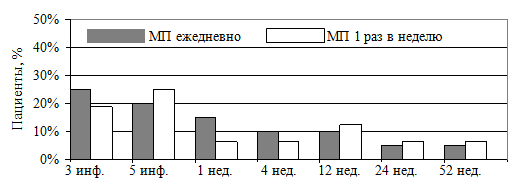
На представленном графике видно, что несмотря на повышение уровня гликемии на фоне проводимой пульс-терапии, последующее наблюдение в течение 1 года не показало повышения риска нарушения углеводного обмена. Таким образом, ни «короткая», ни «длинная» схема пульс-терапии не сопровождается долгосрочным отрицательным влиянием на углеводный обмен. Тем не менее, в период проведения пульс-терапии должен осуществляться контроль гликемии.
Обсуждение
В литературе нам не удалось найти данных о частоте развития нарушения углеводного обмена на фоне терапии ГК при ЭОП. Очевидно, что при пероральном приеме удельный вес всех побочных эффектов составляет 35% случаев, и 23% при использовании пульс-терапии.30 В настоящее временя существуют различные режимы внутривенного введения ГК, но преимуществ какого-либо из них пока не доказано. Недавно проведенное исследование по сравнению различных доз метилпреднизолона (2,25 гр. vs 4,98 гр. vs 7,47 гр.) показало, что средние дозы препарата предпочтительно использовать при ЭОП средней тяжести, тогда как более высокие дозы, в связи с более быстрым достижением положительного эффекта и более частым развитием побочных эффектов, — при тяжелой ЭОП.31
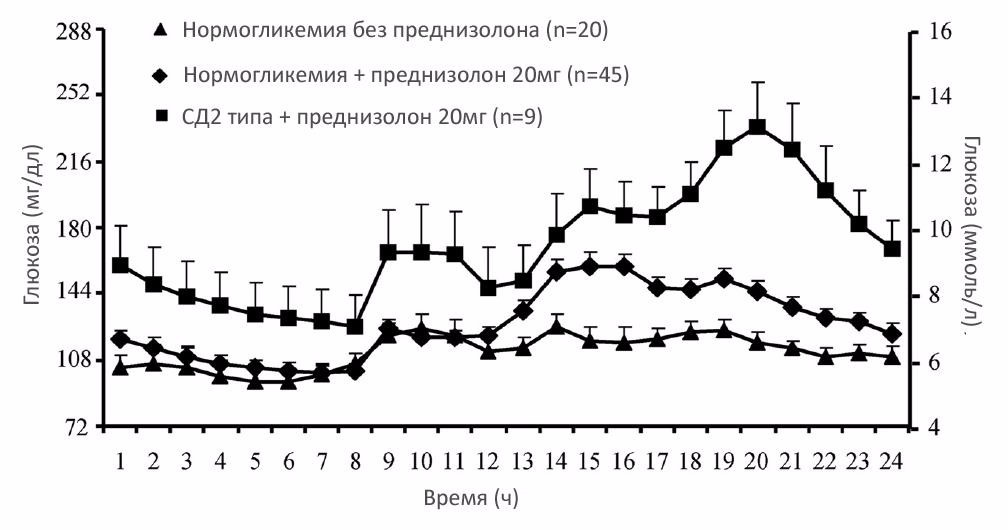
Все чаще и чаще при ЭОП пульс-терапия проводится в течение длительного периода, иногда с назначение преднизолона после ее окончания. Так, в странах Европы при ЭОП метилпреднизолон вводится в дозе 500 мг 1 раз в неделю в течение 6 недель с последующим переходом на 250 мг 1 раз в неделю в течение следующих 6 недель.32 Несмотря на очевидные выводы о большей безопасности пульс-терапии, до сих пор многие пациенты с ЭОП в качестве терапии первого выбора получают таблетированные ГК. В связи с чем, интересны результаты недавно проведенного исследования с участием пациентов, получающих терапию преднизолоном по поводу ХОБЛ, в котором, в отличие от нашего исследования, оценивалась суточная динамика уровня гликемии.33 Как и ожидалось, уровень гликемии на фоне терапии ГК оказался выше у пациентов с СД2 типа. Любопытно, что при приеме преднизолона в утренние часы уровень гликемии достигал своего пика через 8 часов. Если же преднизолон принимался днем, то пик гипергликемии наступал через 5 часов (рис. 8). Таким образом, вне зависимости от времени приема преднизолона пик гипергликемии развивался в одно и тоже время.33
Рисунок 8. Суточная динамика уровня гликемии на фоне терапии ГК33

Эти данные имеют важное клиническое значение: во-первых, контроль уровня гликемии у пациентов, получающих преднизолон, должен проводиться после обеда, до и после ужина, поскольку оценка уровня гликемии в утренние часы малоинформативна для оценки стероид-индуцированной гипергликемии. Во-вторых, сахароснижающая терапия должна назначаться именно в этот период времени – между полуднем и 0.00 ночи.
В 2013г. были опубликованы рекомендации по диагностике и лечению побочных эффектов терапии ГК, в том числе и вторичного сахарного диабета, индуцированного приемом ГК.34 Для диагностики диабета рекомендовано использовать все те же критерии, что и для диагностики диабета любого генеза. Однако считается, что более чувствительным тестом в этом случае является ОГТТ, и менее чувствительным – гликированный гемоглобин, гликемия натощак и случайно измеренный уровень гликемии. Контроль гликемии рекомендуется осуществлять в течение первых 8 часов после начала приема преднизолона. Канадская диабетологическая ассоциация предлагает проводить контроль гликемии через 48 часов после начала терапии ГК. 35
Таким образом, все исследователи сходятся во мнении, что у пациентов, получающих ГК, вне зависимости от типа ГК или длительности терапии, необходимо проводить контроль углеводного обмена. Однако по срокам проведения контроля единого мнения нет. Поскольку определенно точно известно, что длительный прием ГК ассоциирован с ингибирующим влиянием на β-клетки, для более точного понимания влияния пульс-терапии МП при ЭОП необходимо проведение клинических исследований с оценкой различных параметров, отражающих влияние ГК на углеводный обмен.
Список литературы
-
Bartalena L., Baldeschi L., Dickinson A., Eckstein A., Kendall-Taylor P., Marcocci C., et al. Consensus Statement of the European Group on Graves’ Orbitopathy (EUGOGO) on Management of Graves’ Orbitopathy // Thyroid 2008;18:3:333-34
6.doi: 10.1089/thy.2007 .0315. -
Насонова В. А. и др. Современные аспекты глюкокортикоидно
й терапии ревматических заболеваний // Пособие для врачей. – М., 2009. – 40 с. -
Яременко О.Б. Глюкокортикоиды в ревматологии: современная номенклатура дозовых режимов и рациональное применение // Украинский Ревматологически
й Журнал 2002;9:20-26 -
Hiromatsu Y. Steroid therapy for Graves’ ophthalmopathy // Nippon Rinsho 2006;64:12: 2279-2285.
-
Kahaly G. J., Pitz S., Hommel G., Dittmar M. Randomized, single-blind trial of intravenous versus oral steroid monotherapy in Graves’ orbitopathy // J. Clin. Endocrinol. Metab. 2005;90:5234-5240. http://dx.doi.or
g/10.1210/jc.200 5-0148 -
Raalte D.H, Ouwens D.M., Diamant M. Novel insights into glucocorticoid-m
ediated diabetogenic effects: towards expansion of therapeutic options? // Eur J Clin Invest. 2009; 39 (2):81–93 doi: 10.1111/j.1365-2 362.2008.02067.x . -
Raalte D.H., Nofrate V., Bunck M.C. et al. Acute and 2-week exposure to prednisolone impair different aspects of b-cell function in healthy men // European Journal of Endocrinology 2010;162:729–735 doi: 10.1530/EJE-09-1
034 -
Hamamdzic D, Duzic E, Sherlock JD, Lanier SM. Regulation of alpha 2-adrenergic receptor expression and signaling in pancreatic beta-cells. Am J Physiol 1995;269:162–71.
- Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005;365:1333–46
. http://dx.doi.or g/10.1016/S0140- 6736(05)61032-X - Ewart HS, Somwar R, Klip A. Dexamethasone stimulates the expression of GLUT1 and GLUT4 proteins via different signaling pathways in L6 skeletal muscle cells. FEBS Lett 1998;421:120–4. http://dx.doi.or
g/10.1016/S0014- 5793(97)01549-4 -
Long W, Barrett EJ, Wei L, Liu Z. Adrenalectomy enhances the insulin sensitivity of muscle protein synthesis. Am J Physiol Endocrinol Metab 2003;284:102–9. doi: 10.1152/ajpendo.
00028.2002 -
Saad MJ, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinos
itol 3-kinase in liver and muscle of dexamethasone-tr eated rats. J Clin Invest 1993;92:2065–72. doi: 10.1172/JCI11680 3. - Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulat
ed recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism 1998;47:3–6. http://dx.doi.or g/10.1016/S0026- 0495(98)90184-6 -
Lofberg E, Gutierrez A, Wernerman J, Anderstam B, Mitch WE, Price SR et al. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur J Clin Invest 2002;32:345–53. doi: 10.1046/j.1365-2
362.2002.00993.x -
Krebs M, Krssak M, Bernroider E et al. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes 2002;51:599–605. doi: 10.2337/diabetes
.51.3.599 -
Ruzzin J, Wagman AS, Jensen J. Glucocorticoid-i
nduced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 2005;48:2119–30. doi: 10.1007/s00125-0 05-1886-0 -
Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 2002;32(Suppl. 3):14–23. DOI: 10.1046/j.1
365-2362.32.s3.3 .x -
Perseghin G, Petersen K, Shulman GI. Cellular mechanism of insulin resistance: potential links with inflammation. Int J Obes Relat Metab Disord 2003;27(Suppl. 3):S6–11.
-
Nielsen MF, Caumo A, Chandramouli V, Schumann WC et al. Impaired basal glucose effectiveness but unaltered fasting glucose release and gluconeogenesis during short-term hypercortisolemi
a in healthy subjects. Am J Physiol Endocrinol Metab2004;286:102–10. DOI: 10.1152/ajpendo. 00566.2002 -
Rooney DP, Neely RD, Cullen C, Ennis CN et al. The effect of cortisol on glucose/glucose-
6-phosphate cycle activity and insulin action. J Clin Endocrinol Metab 2013;77:1180–3. DOI: http://dx.doi.or g/10.1210/jcem.7 7.5.8077310 -
Jin JY, Dubois DC, Almon RR, Jusko WJ. Receptor/gene-me
diated pharmacodynamic effects of methylprednisolo ne on phosphoenolpyruv ate carboxykinase regulation in rat liver. J Pharmacol Exp Ther 2004;309:328–39. doi:10.1124/jpet.103 .061515 -
Sasaki K, Cripe TP, Koch SR, Andreone TL, Petersen DD, Beale EG et al. Multihormonal regulation of phosphoenolpyruv
ate carboxykinase gene transcription. The dominant role of insulin. J Biol Chem 1984;259:15242–5 1. -
Vander Kooi BT, Onuma H, Oeser JK, Svitek CA, Allen SR et al. The glucose-6-phosph
atase catalytic subunit gene promoter contains both positive and negative glucocorticoid response elements. Mol Endocrinol 2005;19:3001–22. DOI: http://dx.doi.or g/10.1210/me.200 4-0497 -
Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol 2007;275:43–61.doi:10.1016/j.mc
e.2007.05.015 -
Kalhan SC, Adam PA. Inhibitory effect of prednisone on insulin secretion in man: model for duplication of blood glucose concentration. J Clin Endocrinol Metab 1975;41:600–10. DOI: http://dx.doi.or
g/10.1210/jcem-4 1-3-600 -
Van Raalte DH, Mari A, Bunck MC et al. Acute and subacute exposure to glucocorticoids differentially impair various aspects of beta-cell function in healthy males (1468-P). 68th Sessions American Diabetes Association. San Francisco, 2008
-
Clore J, Thurby-Hay L. Glucocorticoid-I
nduced Hyperglycemia // Endocrine Practice 2009;15 (5):469-474 doi: 10.4158/EP08331. RAR. -
Gurwitz J.H. Bohn R.L. et al. Glucocorticoids and the Risk for Initiation of Hypoglycemic Therapy // Arch Intern Med. 1994;154:97-101 doi:10.1001/arch
inte.1994.004200 10131015. -
Триголосова И.В. Нарушения углеводного обмена и контринсулярные гормоны // Опухоли эндокринной системы с различной гормональной активностью. Материалы областной практической конференции 2015:15-17
-
Marcocci C., Bartalena L., Tanda M. L., et al. Comparison of the effectiveness and tolerability of intravenous or oral glucocorticoids associated with orbital radiotherapy in the management of severe Graves’ ophthalmopathy: results of a prospective, single-blind, randomized study // J. Clin. Endocrinol. Metab. 2001;86:3562-356
7. DOI: http://dx.doi.or g/10.1210/jcem.8 6.8.7737 -
Bartalena L., Krassas G.E., at al. Efficacy and safety of three different cumulative doses of intravenous methylprednisolo
ne for moderate to severe and active Graves’ orbitopathy // J. Clin. Endocrinol. Metab. 2012;97 (12):4454-4463 doi: 10.1210/jc.2012- 2389 -
Zang S., Ponto K. A., Kahaly G. J. Intravenous glucocorticoids for Graves’ orbitopathy: efficacy and morbidity // J. Clin. Endocrinol. Metab. 2011;96 (2):320-332. DOI: http://dx.doi.or
g/10.1210/jc.201 0-1962 -
Morton G. Burt, Gregory W. Roberts, Norma R. Aguilar-Loza, Continuous Monitoring of Circadian Glycemic Patterns in Patients Receiving Prednisolone for COPD // J Clin Endocrinol Metab 2011;96:1789–179
6 doi: 10.1210/jc.2010- 2729 -
Liu D, Ahmet A., Ward L. et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy // Allergy, Asthma & Clinical Immunology 2013;9:30 doi: 10.1186/1710-149
2-9-30. -
Canadian Diabetes Association Clinical Practice Guidelines Expert Committee: Canadian Diabetes Association 2013 clinical practice guidelines for the prevention and management of diabetes in Canada // Can J Diabetes 2013, 37(Suppl 1):S1–S212.