


Макрофаги, ассоциированные с опухолями (ОАМ) играют значимую роль в развитии онкологических заболеваний.
- Рак у человека вызывается мутацией гена рецептора PPARg с потерей его функции, но развитие опухоли могут сдерживать агонисты PPARg класса тиазолидиндионов (ТЗД), в том числе росиглитазон.
- Неясным остается, каким именно образом макрофаги реализует противоопухолевую функцию PPARg.
- В эксперименте на мышах обнаружено, что устранение PPARg не только негативно влияет на развитие опухолей молочной железы, но и ослабляет противоопухолевые эффекты росиглитазона.
- В качестве новой мишени для PPARg был определен Gpr132 (G-белок, соединенный рецептором 132), экспрессия которого увеличивается в макрофагах при потере функции PPARg и уменьшается при активации PPARg соответственно.
- Gpr132 в макрофагах реализует провоспалительные и проопухолевые эффекты.
- Генетическая делеция Gpr132 замедляет воспаление и прогрессирование рака, но в то же время нивелирует противоопухолевый эффект PPARg и росиглитазона.
- Фармакологическое ингибирование Gpr132 значительно препятствует малигнизации опухолей молочной железы.
Резюмируя, необходимо отметить:
- ведущую роль PPARg и Gpr132 в качестве модуляторов активности ОАМ;
- потенциально новые мишени в терапии рака;
- препараты класса тиазолидиндионов как важнейшие медиаторы антиопухолевой активности.
Справочная информация
- Иммунная система может оказывать влияние на опухолевый процесс двояким образом: как стимулировать, так и сдерживать рост опухолей.
- Макрофаги создают воспалительное микроокружение вокруг опухоли, что способствует распространению раковых клеток.
- Независимые наблюдения и эксперименты доказали, что протеин PPARg (рецептор активаторов пролиферации пероксисом гамма) способен оказывать супрессивное действие на рост опухолей.
- Препараты группы тиазолидиндионов, которые традиционно использовались в терапии сахарного диабета 2 типа, активируют PPARg и за счет этого реализуют противоопухолевый эффект. Однако данный механизм до сих пор не изучен досконально.
Существует гипотеза (Cheng et al.) о том, что PPARg и ТЗД ингибируют активность провоспалительных макрофагов, которые участвуют в развитии опухолей.
- Чтобы подтвердить данное предположение, использовали мышей, которым при помощи генной инженерии полностью устранили PPARg.
- У этих мышей отмечался более высокий уровень развития рака молочной железы (РМЖ) в сравнении с обычными особями.
- Более того, у искусственно выведенных мышей, опухоли молочной железы не уменьшались при использовании терапии ТЗД, в то время как для прочих мышей терапия была эффективной.
Еще одно открытие заключается в том, что PPARg препятствует продукции протеина Gpr132 макрофагами.
- Gpr132 обладает провоспалительной активностью и способствует росту опухолевых клеток в случае РМЖ.
- У мышей, неспособных к продукции Gpr132, наблюдались вялотекущие процессы воспаления и торможение опухолевой прогрессии.
- Препараты, ингибирующие активность Gpr132 у обычных мышей, также показали эффективное снижение прогрессии опухолей.
Последующие эксперименты должны быть направлены на изучение механизма, посредством которого Gpr132, продуцируемый макрофагами, воздействует на опухолевые клетки. В дальнейшем, применение фармакологических средств – ингибиторов Gpr132, может значительно повлиять на эффективность терапии рака.
Введение
Колоссальную терапевтическую значимость для борьбы с раком несет в себе ответ на фундаментальный вопрос, каким образом активность иммунных клеток в опухолевом микроокружении связана со степенью злокачественности рака.
Очевидные доказательства свидетельствую в пользу функциональной связи между воспалением и развитием рака.
- Очаги хронического воспаления, усиливающие канцерогенез, обнаруживаются более чем в 15% случаев возникновения рака.
- ОАМ, как ключевой участник воспалительного процесса и прогрессии рака, напрямую коррелирует с ухудшением прогноза.
К примеру, чрезмерная экспрессия колониестимулирующего фактора 1 (КСФ1) приводит к ускорению опухолевой прогрессии у мышей и у людей. Более того, ОАМ также регулирует ответ на терапию.
Несмотря на то, что многочисленные клинические исследования и экспериментальные модели на мышах поддерживают теорию о том, что макрофаги главным образом обладают проопухолевой активностью, противоопухолевые эффекты также обнаружены у некоторых подтипов макрофагов.
Эти данные свидетельствуют в пользу того, что роль, отведенная макрофагам в канцерогенезе, неоднозначна и изменчива в зависимости от ситуации.
PPARg (рецептор активаторов пролиферации пероксисом гамма) — ядерный рецептор и фактор транскрипции, регулирующий бессчетное число физиологических процессов. Мутацию PPARg с потерей его функции связывали с развитием рака у человека.
- Синтетические агонисты PPARg, такие как препараты тиазолидиндионов (противодиабетические средства росиглитазон и пиоглитазон), применялись с целью сдерживания малигнизации опухолей. Несмотря на это, к настоящему моменту клинические испытания не пришли к общему заключению относительно влияния препаратов группы тиазолидиндионов на исход рака.
- Анализ рандомизированных клинических испытаний демонстрирует интересную статистику: случаи малигнизации опухолевых образований значительно ниже в группе пациентов, получающих лечение росиглитазоном, по сравнению с контрольной группой, однако, росиглитазон не являлся модифицирующим фактором в возникновении рака.
Полученные данные не только подтверждают противоопухолевую активность росиглитазона, но также дают основания предполагать, что препараты группы тиазолидиндионов в большей степени замедляют развитие опухоли, нежели препятствуют ее появлению.
- Важно отметить, что эпидемиологические исследования обнаруживают двунаправленную взаимосвязь между сахарным диабетом и раком: диабет (преимущественно СД 2 типа) коррелирует с более высоким риском развития рака, включая рак молочной железы;
- в то же время, 8-18% рака впервые диагностируются у пациентов с диабетом, и у таких пациентов (с предсуществующим диабетом) выживаемость после хирургического лечения ниже.
Таким образом, понимание механизма воздействия противодиабетических препаратов на развитие и прогрессирование рака имеет первостепенную важность для борьбы с обоими заболеваниями.
В центре внимания предыдущих исследований в большей степени были эффекты PPARg, оказываемые на опухолевые клетки.
- Тиазолидиндионы способствуют окончательной дифференцировке клеток опухоли молочной железы, устраняют пролиферацию и триггерное накопление липидов клеток липосаркомы.
- Еще в одном исследовании обнаружено, что антипролиферативный эффект тиазолизиндионов связан с метаболическим сдвигом в клетках опухолей легкого и молочной железы.
Несмотря на все полученные данные, роль PPARg в макрофагах в плане воздействия на прогрессию рака, остается до конца не изученной.
- Более того, предыдущие исследования во многом опирались на использование лигандов PPARg, способных активировать PPARg-независимые и/или физиологически неподходящие эффекты;
- генетический разбор специфических функций PPARg для каждого типа клеток в опухолевом окружении in vivo не проводился.
Ранее было доложено о том, что у самок мышей с делецией PPARg в гемопоэтических и эндотелиальных клетках, воспаление развивалось в лактирующей молочной железе. Это привело к продукции молока, содержащего факторы воспаления, что послужило причиной системного воспаления у новорожденных особей и манифестировало в виде временной потери меха.
- Это наблюдение дает повод предполагать, что PPARg играет противовоспалительную роль в макрофагах и в молочной железе, что может иметь значение в развитии рака.
Возникает закономерная гипотеза о том, что PPARg в макрофагах препятствует развитию рака молочной железы путем подавления воспаления.
- При помощи различных подходов, увеличивающих и подавляющих активность соответствующих генов, в серии генетических и фармакологических экспериментов in vitro и in vivo, были обнаружены онкосупрессивные функции PPARg относительно прогрессирования рака молочной железы.
- К тому же, PPARg показал себя ключевым медиатором противоопухолевых эффектов росиглитазона за счет репрессинга новой мишени — Gpr132.
Результаты
Делеция макрофагального PPARg усиливает опухолевый рост in vivo
Путем выведения от мышей, имеющих аллель фланкированных loxP-сайтов (флоксированный ген) и трансгенных по Cre-рекомбиназе (активируется Tie2Cre или Lysozyme-Cre промотором), были получены особи с нокаутом по PPARg в макрофагах (mf-g-KO).
- Tie2Cre устраняет PPARg в гемопоэтических и эндотелиальных клетках.
- LyzCre удаляет PPARg в миелоидном ростке клеток.
Две эти модели mf-g-KO взаимно дополняют друг друга:
- у Lyz-g-KO мышей реализована более специфичная делеция PPARg;
- Tie2-g-KO мыши имеют более полную (89%) делецию макрофагального PPARg, в сравнении с Lyz-g-KO (79%);
- сравнению в рамках обеих моделей подвергались однопомётные животные PPARgflox/flox;Cretg/+ KO и PPARgflox/flox;Cre+/+
С целью определения эффектов делеции макрофагальных PPARg для развития РМЖ, женским особям мышей были выполнены инъекции опухолевых клеток EO771 в жировую ткань молочной железы от линии C57BL/6J-совместимых мышей. Рост опухоли контролировался по ее размерам.
- Сравнивая однопомётных мышей, можно отметить, что и Tie2-g-KO, и Lyz-g-KO особи показали в равной степени усиление роста и увеличение объема опухоли (рис.1A–B).
{kind=link}
Полученные данные говорят о том, что наблюдаемый проопухолевый эффект в большей степени вызван делецией PPARg в миелоидных клетках и в макрофагах в частности.
- При окрашивании для выявления маркеров Ki67 и фосфо-гистонов H3 (PH3) на срезах опухоли обнаружилось значительное повышение клеточной пролиферации у mf-g-KO мышей (рис.1C–D).
Чтобы смоделировать иную модель развития рака, мышам другой C57BL/6-совместимой линии были привиты клетки (Py230), извлеченные из спорадических опухолей молочной железы у линии трансгенных мышей C57BL/6 MMTV-PyMT.
- Инъекции клеток Py230 в жировую ткань молочной железы продемонстрировали обострение опухолевой прогрессии у mf-g-KO мышей в сравнении с контрольной группой (рис.1A).
Вышеописанные эксперименты подтверждают способность PPARg в макрофагах ингибировать опухолевый рост in vivo.
Делеция PPARg в макрофагах вызывает избыток опухоль-ассоциированных макрофагов in vivo
Сравнение экспрессии генов в тканях опухолей, клетках костного мозга и клетках селезенки, взятых от мышей mf-g-KO и контрольной группы, показали, что:
- экспрессия провоспалительных генов (в том числе COX-2, MMP9, MCP-1, TNFa and IL-1b (рис.1E–G)) выше в клетках и тканях, лишенных PPARg;
- экспрессия маркеров макрофагов типа M2 (аргиназа 1 (рис.1B)) была понижена.
Эти наблюдения согласуются с данными от нескольких лабораторий: в условиях недостатка PPARg активируются провоспалительные макрофаги, но истощается росток M2.
- Макрофагальная инфильтрация опухоли является четким индикатором злокачественности и неблагоприятного прогноза.
- Иммунофлуоресцентное окрашивание с CD11b и F4/80 выявляет снижение жизнеспособности ОАМ в обеих моделях (Lyz-g-KO и Tie2-g-KO) в сравнении с контрольной группой (рис. 1H).
- Это согласуется с полученными ранее данными о том, что макрофаги, лишенные PPARg, склонны к миграции и экспрессии CCR2 (хемокиновый рецептор 2 типа), тогда как лечение ТЗД подавляет миграцию макрофагов и экспрессию CCR2.
- В соответствии с информацией о том, что агонисты PPARg ингибируют ангиогенез, было обнаружено, что количество сосудов на срезах опухолей выше у Tie2-g-KO мышей, и остается неизменным у Lyz-g-KO (рис.1E–F).
- Таким образом, устранение PPARg в макрофагах само по себе способствует усилению опухолевого роста вне зависимости от изменений в процессе ангиогенеза.
Как итог данного этапа получено заключение: устранение PPARg в макрофаге влияет на число и функцию ОАМ в процессе формирования провоспалительного опухолевого микроокружения.
Макрофаги, лишенные PPARg, активируют пролиферацию опухолевых клеток in vitro
Серия экспериментов с культурой опухолевых клеток и макрофагами была проведена in vitro (рис. 2A), чтобы определить, могут ли макрофаги, лишенные PPARg, влиять на активность опухолевых клеток в отсутствии других компонентов опухолевого микроокружения, таких как фибробласты и экстрацеллюлярный матрикс.
{kind=link}
- Макрофаги мышей, полученные от клеток-предшественников из костного мозга или селезенки, соединили в единую культуру с линией клеток РМЖ человека MDA-MB-231, маркированных люциферазой (1833 клетки).
- Таким образом, оказался возможен специфический количественный анализ уровня пролиферации опухолевых клеток.
- Результаты продемонстрировали значимое увеличение пролиферации опухолевых клеток за счет макрофагов, лишенных PPARg, в сравнении с контролем (рис. 2B).
- В соответствии с последним наблюдением, культура, объединяющая макрофаги без PPARg и опухолевые клетки, также продемонстрировала увеличение формирования колоний опухолевых клеток.
Поскольку макрофаги мышей и опухолевые клетки человека относятся к разным видам, экспрессия мРНК в двух данных типах клеток в их совместной культуре может быть разделена посредством количественной ПЦР с видоспецифичными праймерами.
Культура, включающая макрофаги без PPARg, показала более высокую экспрессию маркеров пролиферации и более низкий уровень экспрессии маркеров апоптоза в сравнении с контрольной группой макрофагов (рис. 2D–E).
В соответствии с наблюдениями in vivo (рис.1) макрофаги, лишенные PPARg, продемонстрировали повышенную экспрессию провоспалительных генов (COX-2, MCP-1, MMP-9) и сниженный уровень маркёров линии макрофагов M2 (аргиназа-1 (рис. 2F).
- Макрофаги без PPARg показали более высокий уровень генов антиапоптоза и более низкие уровни генов, запускающих апоптоз, что увеличивает выживаемость.
- Более того, макрофаги, лишенные PPARg склонны к более активной пролиферации, измеренной посредством специфических индикаторов метаболизма ATP (рис. 2H) или MTT.
Результаты исследования in vitro подтверждают наблюдения in vivo: более высокой численности макрофагов, лишенных PPARg и их провоспалительных эффектов достаточно для стимуляции опухолевой прогрессии.
Активация PPARg в макрофагах посредством росиглитазона сдерживает пролиферацию опухолевых клеток in vitro
В качестве дополнения к эксперименту с генетически-опосредованной потерей функции PPARg, следующим шагом стал эксперимент с фармакологическим эффектом усиления функции, чтобы оценить влияние макрофагальных PPARg (активированных росиглитазоном) на опухолевые клетки.
- Макрофаги мышей одной группы были предварительно обработаны росиглитазоном, другой – инертным веществом;
- росиглитазон был удален прежде, чем опухолевые клетки человека добавили к культуре (рис. 2A).
Результаты показали, что рост опухолевых клеток значительно снизился в культуре с макрофагами, обработанными росиглитазоном, в противоположность к группе сравнения (рис. 2I).
- Важно отметить, что данный эффект росиглитазона является зависимым от макрофагальных PPARg. Культура с макрофагами, лишенными PPARg, не показала различий при обработке росиглитазоном или инертным веществом (рис. 2I).
В качестве положительного контроля эффекта индукции PPARg росиглитазоном, мишенью которого является ген LXRa, исследовались макрофаги мышей WT (wild type), но не g-KO (рис.2).
Объединяя все данные, можно утверждать, что активация PPARg в макрофагах эндогенными равно как и синтетическими агонистами, вызывает супрессию опухолевого роста.
PPARg в макрофаге является ключевым медиатором противоопухолевой активности росиглитазона in vivo
При оценке функциональной значимости макрофагальных PPARg для фармакологических эффектов росиглитазона, на мышах mf-g-KO и однопометных особях применили росиглитазон и инертное вещество через 4 дня после инъекции опухолевых клеток.
- Результаты показали, что способности росиглитазона оказывать супрессивное воздействие на рост клеток РМЖ значительно ослабли у мышей группы mf-g-KO (рис. 2J).
- В соответствии с этим наблюдением, способность росиглитазона уменьшать количество ОАМ так же не проявилось у mf-g-KO мышей (рис.2).
Данные демонстрируют, что макрофаг является необходимым субстратом для реализации противоопухолевого эффекта росиглитазона.
PPARg в макрофагах снижает экспрессию Gpr132
Чтобы понимать, каким образом PPARg изменяет транскрипционную программу макрофага для контроля над пролиферацией опухолевых клеток, следующим шагом необходимо обозначить ключевые гены – мишени PPARg.
- Эксперименты выявили, что пролиферация опухолевой клетки может быть значительно повышена путем совмещения культур клеток с макрофагами, лишенными PPARg (рис. 3A–B), но не в условиях питательной среды PPARg-дефицитных макрофагов.
- Это объясняется тем, что необходим физический контакт между макрофагами и опухолевыми клетками, следовательно, ключевой модулирующий опухоль PPARg-целевой ген в макрофаге кодирует мембранный протеин.
- Путем анализа существующей базы данных соответствующих исследований были отобраны наиболее вероятные мембранные протеины, которые могут регулироваться посредством PPARg:
- G-белок, соединенный рецептором 132 (G2A), показал постоянную и значительную положительную регуляцию в макрофагах, дефицитных по PPARg в сравнении с контрольной группой макрофагов (WT);
- 11 прочих отобранных мембранных белков оставались неизменными (рис.3);
- Таким образом, главным вопросом стало, является ли Gpr132 функциональной мишенью для PPARg в макрофаге.
{kind=link}
Прежде Gpr132 описывался как стресс-индуцируемый трансмембранный рецептор с семью каналами, который функционирует на контрольной точке G2/M клеточного цикла и модулирует иммунную функцию клетки.
- Gpr132 преимущественно экспрессируют клетки гемопоэтического ряда, а также макрофаги. Экспрессия Gpr132 в опухолевых клетках и прочих тканях минимальна, что подтверждает предположение о специфической функции этого протеина в макрофагах (рис. 3C–D).
- Экспресиия Gpr132 в PPARg-дефицитных макрофагах значительно превышает таковую в контрольной группе, как в изолированной культуре макрофагов, так и совместно с опухолевыми клетками (рис. 3E–F).
- В соответствии с этими наблюдениями, активация PPARg росиглитазоном, снижала экспрессию Gpr132 в макрофагах группы WT, но не изменяла ее уровень у PPARg-дефицитных макрофагах (рис. 3G).
Наблюдения подтверждают, что PPARg угнетают экспрессию Gpr132.
PPARg встраивается в промотор Gpr132 и угнетает его транскрипцию
- Область промотора Gpr132 (0.5 kb and 1kb) перенесли в вектор репортерного гена люциферазы;
- временный перенос генов и последующий анализ по гену-репортеру показал, что устранение обоих участков промотора Gpr132 редуцируется посредством котрансфекции PPARg и, в дальнейшем, применением росиглитазона (рис. 3H).
PPARg ингибирует промотор Gpr132 через критические элементы в пределах 500 пар оснований вверх относительно начала сайта транскрипции Gpr132. Был определен ответный элемент в PPARg (PPRE) в половине сайта данной области (-188: CATCCGAGCAAGGTCAGAC).
- Анализ хроматин-иммунопреципитации обнаружил, что PPARg может связываться с проксимальным эндогенным промотором Gpr132 в макрофаге, но не выше отрицательной области управления (рис. 3I);
- Росиглитазон не оказывает влияния на эту связь, более того, анализ хроматин-иммунопреципитации определил, что H3K9Ac (активный транскрипционный гистон в стартовом сайте транскрипции Gpr132) стимулируется выключением PPARg и выключается росиглитазоном (в соответствии с воздействием росиглитазона на PPARg (рис. 3J)).
Таким образом, PPARg, при активации эндогенным или синтетическим лигандом, напрямую угнетает транскрипцию Gpr132 в макрофаге .
Gpr132 в макрофагах человека репрессируется PPARg и коррелирует с РМЖ у человека
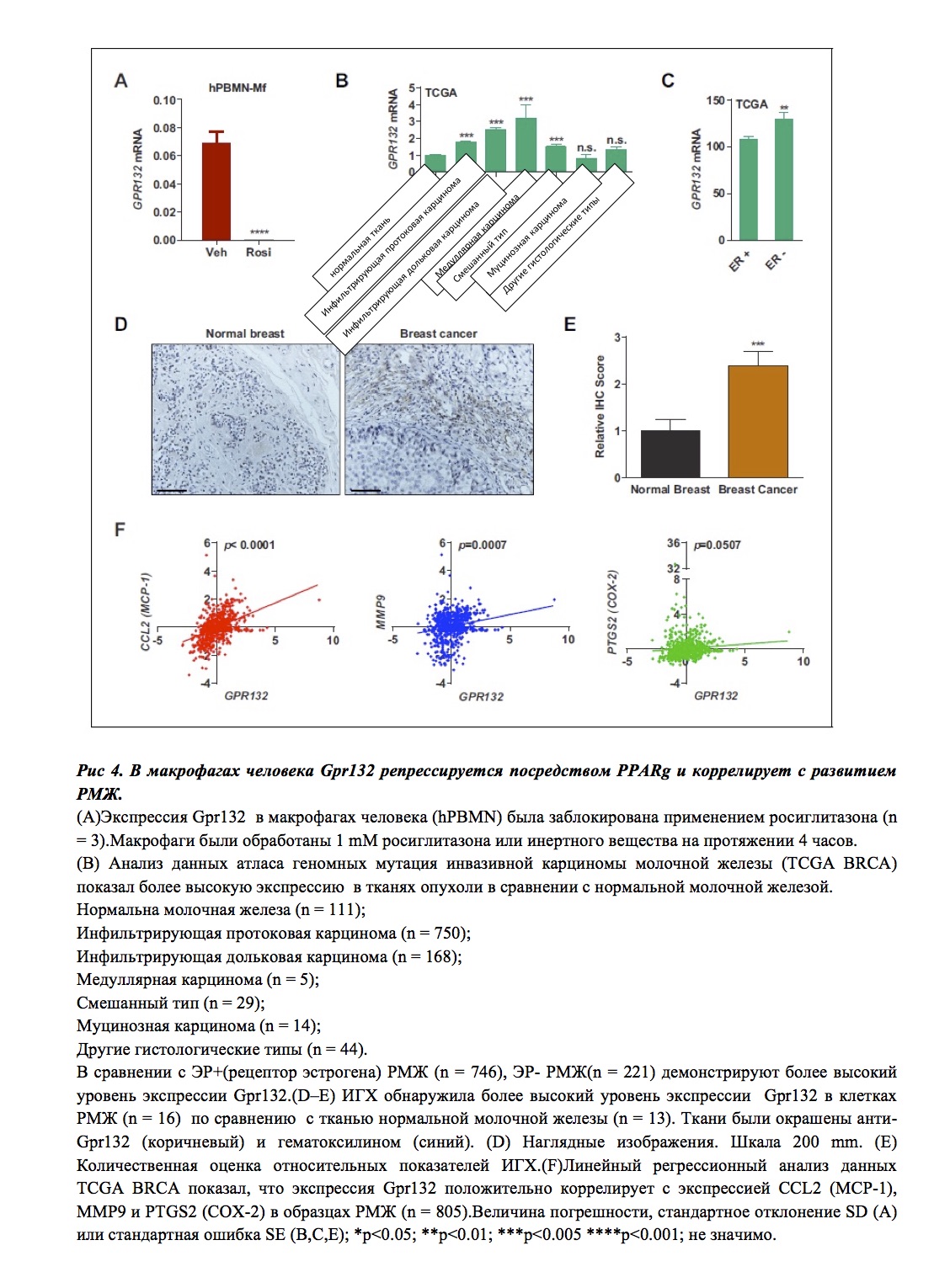
Экспрессия Gpr132 в макрофагах, полученных от мононуклеарных клеток периферической крови (hPBMN) также угнетается росиглитазоном (рис. 4A). Это означает, что репрессинг Gpr132 посредством PPARg сохраняется эволюционно, и результаты, полученные в эксперименте на мышах, можно применить к физиологии и болезням человека.
{kind=link}
- Исследование инвазивной карциномы молочной железы путем секвенирования РНК и при помощи клинических данных, а также посредством данных атласа генетических мутаций, ассоциированных с онкологическими заболеваниями (TCGA) подтвердили значимую роль Gpr132 в развитии РМЖ.
- Поскольку Gpr132 в большом количестве экспрессируется в человеческих макрофагах (рис. 4A), но отсутствует в клетках РМЖ (рис. 3D), присутствие Gpr132 в опухоли обусловлено наличием гемопоэтических клеток (и макрофагов в частности) в опухолевом микроокружении.
- Сравнивая ткани нормальной молочной железы и ткани, пораженные опухолью, можно отметить значительно большую экспрессию Gpr132 в последних (рис. 4B).
- Наиболее агрессивные эстроген-негативные опухоли МЖ также показали высокую экспрессию Gpr132 (рис. 4C).
- Иммуногистохимический анализ подтвердил, что ткани МЖ, пораженной опухолевым процессом, экспрессируют значительно более высокий уровень Gpr132 в сравнении с нормальными тканями МЖ (рис. 4D–E).
- Линейный регрессивный анализ обнаружил, что высокий уровень Gpr132 коррелирует с высокой экспрессией провосполительных маркеров (включая CCL2 (MCP-1), MMP9 и PTGS2 (COX-2)) при поражении тканей РМЖ (рис. 4F).
Можно предположить, что Gpr132 играет роль в развитии воспаления и прогрессировании опухолевого процесса.
Gpr132 в макрофагах способствует пролиферации опухолевых клеток in vitro
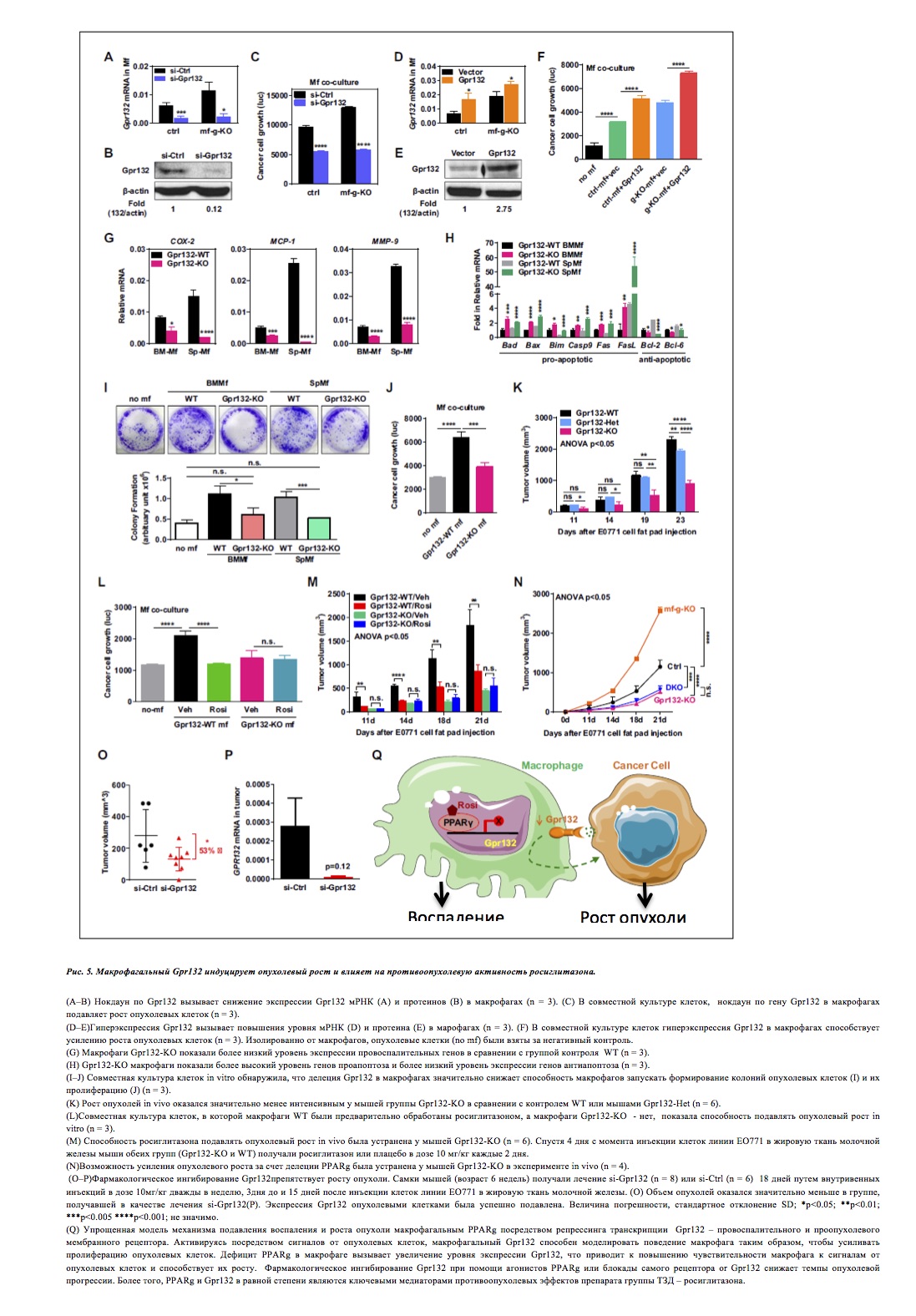
Выключение Gpr132 в макрофагах значительно снижает рост опухолевых клеток (рис. 5A–C) и наоборот, высокий уровень экспрессии Gpr132 стимулирует рост опухолевых клеток (рис. 5D–F).
{kind=link}
- При помощи мышей группы Gpr132-KO были получены анти- Gpr132 антитела (рис.5).
- Было проведено сравнение макрофагов, полученных из костного мозга или селезенки мышей группы Gpr132-KO и однопомётных мышей контрольной (WT) группы.
- Генетический анализ выявил, что макрофаги Gpr132-KO экспрессируют фенотип, противоположный таковому у PPARg-дефицитных макрофагов, с меньшим количеством провоспалительных генов (рис. 5G), более высоким уровнем экспрессии генов апоптоза и с пониженным для генов анти-апоптоза (рис.5H) в сравнении с контрольной группой.
- Таким образом, in vitro совместная культура макрофагов и опухолевых клеток обнаружила значительное снижение возможности макрофагов группы Gpr132-KO влиять на формирование колоний опухолевых клеток и их рост (рис. 5I–J).
Gpr132 повышает выживаемость макрофагов и усиливает воспаление, а активированные Gpr132 в PPARg-дефицитных макрофагах могут брать на себя функцию стимулирования опухолевой прогрессии.
Полное выключение Gpr132 демонстрирует уменьшение опухолевого роста в эксперименте на мышах
Мышам типа Gpr132-KO и контрольной группе (WT) произвели инъекцию клеток РМЖ EO771.
- При этом Gpr132 был полностью удален из макрофагов, костного мозга, тимуса, селезенки. Опухолевые клетки не экспрессируют Gpr132 (рис. 3C–D).
- Предыдущие эксперименты показали, что у мышей группы Gpr132-KO сохраняется нормальная линия дифференциации Т и B лимфоцитов вплоть до 1го года, а затем начинается их патологическая трансформация с прогрессирующим увеличением лимфоидных органов.
- В этой связи настоящий эксперимент проводился на молодых особях до момента, когда дефекты лимфоидной ткани могли бы потенциально повлиять на опухолевую прогрессию.
- В сравнении гетерозигот Gpr132-KO и WT, мыши Gpr132-KO показали значительно меньшее влияние на опухолевый рост. Аналогичный результат был выявлен и для более поздней стадии (рис. 5K).
В совокупности полученные данные свидетельствуют в пользу того, что Gpr132 стимулирует опухолевый рост, следовательно, ингибируя Gpr132 можно сдерживать опухолевую прогрессию.
Gpr132 в макрофаге влияет на PPARg и на эффекты, оказываемые росиглитазоном
Культура, совмещающая, макрофаги и опухолевые клетки и мыши, имеющие опухоли, получали росиглитазон или инертное вещество в качестве препарата контроля.
- Предварительная обработка культуры Gpr132-KO макрофагов росиглитазоном (до добавления опухолевых клеток), не показала снижения уровня пролиферации опухолевых клеток (рис. 5L).
- Репрессия Gpr132 посредством PPARg в макрофагах оказывается важным звеном в каскаде противоопухолевых эффектов росиглитазона.
Для применения противоположной стратегии (снижения функции PPARg), были скрещены Gpr132-KO и mf-g-KO линии мышей, в результате чего были выведены mf-g/Gpr132 DKO мыши.
- Эксперимент, аналогичный вышеописанному, у данной группы мышей продемонстрировал, что делеция Gpr132 снижает возможность PPARg-дефицитных макрофагов усугублять опухолевую прогрессию, т.к. данные мыши имели такой же объем опухолей, как и линия Gpr132-KO (рис. 5N).
Таким образом, Gpr132 подтверждает свою роль в качестве необходимого субстрата PPARg в макрофаге для регуляции прогрессии РМЖ.
Фармакологическое ингибирование Gpr132 сдерживает рост опухоли
Поскольку предшественники макрофагов находятся в гемопоэтических тканях (кровь, костный мозг, селезенка), Gpr132 в них можно эффективно ингибировать посредством интерферирующей РНК (siRNA).
- Самок мышей WT лечили посредством si-Gpr132 или si-Ctrl на протяжении 18 дней (3 дня до подсадки опухолевых клеток и 15 дней после) путем внутривенных инъекций (по 10мг дважды в неделю).
- Результаты показали, что si-Gpr132 способствует значительному уменьшению объема опухоли в сравнении с si-Ctrl (рис. 5O). Масса тела осталась неизменной у группы si-Gpr132.
Результаты поддерживают теорию о том, что ингибирование Gpr132 может реализоваться в новейшую стратегию борьбы с опухолевыми заболеваниями.
Обсуждение
PPARg и препараты группы тиазолидиндионов имеют каждый свою нишу в физиологии и лечении заболеваний человека, необходимо выяснить механизмы их воздействия на опухолевый процесс.
В данной работе были раскрыты неизвестные ранее и ключевые пути подавления опухолевой прогрессии посредством PPARg в макрофагах, а также раскрыта роль PPARg в качестве регулятора эффектов росиглитазона (рис. 5Q).
Активация PPARg в макрофаге препятствует реализации воспалительной программы посредством репрессинга транскрипции целевого гена Gpr132, который в свою очередь является провоспалительным мембранным рецептором (рис. 5Q).
Рост опухоли ингибируется при условиях:
- низкого уровня Gpr132;
- делеции или выключения Gpr132;
- активации PPARg росиглитазоном.
Если уровень экспрессии Gpr132 высокий в результате инактивации PPARg в макрофаге, рост опухоли ускоряется.
- Важно отметить, что делеция Gpr132 отменяет эффекты PPARg или росиглитазона на опухолевую прогрессиию, что подчеркивает исключительное значение Gpr132 в качестве медиатора функций PPARg в макрофаге, а следовательно и в качестве инструмента воздействия на развитие опухоли.
PPARg и Gpr132 являются ключевыми игроками во взаимодействии ОАМ с опухолью, раскрывающими механизмы взаимодействия макрофагов и опухолевых клеток и формирование злокачественных новообразований.
Хотя эксперимент с совместной культурой клеток демонстрирует, что необходим физический контакт с опухолевой клеткой, чтобы макрофаг мог реализовать эффекты через PPARg, не исключены и другие пути взаимодействия.
- Непосредственная близость (но не прямой контакт) макрофага и опухолевой клетки необходима для того, чтобы возрастающая экспрессия Gpr132 соответствовала градиенту сигнала от клеток опухоли. Эти данные подтверждены предыдущими исследованиями, которые выявили, что Gpr132 чувствителен к уровню pH и липидов, что позволяет ему регулировать направление миграции иммунокомпетентных клеток.
- Путем активации посредством сигналов от опухоли, Gpr132 способен модулировать внутриклеточные сигналы в макрофаге и воздействовать на другие мишени, что приводит к повышению пролиферации опухолевых клеток (рис5Q).
Данные исследования – первые шаги в совершенно новом направлении на пути к понимаю того, как Gpr132 преобразует сигналы от опухолевых клеток и усиливает степень злокачественности новообразования.
- Макрофаги фенотипов М1 и М2 потенциально обладают проопухолевой активностью.
- Последние литературные данные поддерживают теорию о том, что воспаление способствует опухолевой прогрессии.
- Эксперименты in vivo и in vitro подтверждают теорию о том, что активация макрофагальных PPARg играет противовоспалительную и противоопухолевую роль, а их делеция способствует развитию воспаления и прогрессированию опухолевого процесса.
- В регуляции проопухолевой активности макрофагов PPARg влияет не только на поведение опухолевых клеток, но также воздействует на противоопухолевые эффекторные лимфоциты и иммунный ответ.
- PPARg реализует свое противоопухолевое воздействие на нескольких типах клеток, в том числе иммунокомпетентных.
- Макрофаги являются едва ли ни единственным видом клеток, необходимым для осуществления супрессивного эффекта ТЗД на опухоль.
Хотя в линии мышей Tie2-g-KO была выполнена более полная делеция PPARg, чем в линии Lyz-g-KO, обе модели показали увеличение роста опухоли и взаимодействие опухоль-макрофаг, фенотип лучше определялся в линии Lyz-g-KO. Возможно, что делеция PPARg в гемопоэтических клетках вне миелоидного ростка оказывает незначительное, но противоположно направленное влияние на рост опухоли у мышей типа Tie2-g-KO в отличие от Lyz-g-KO.
PPARg может влиять на многие гены в макрофаге и многие лиганды помимо Gpr132 могут также играть роль в реализации противоопухолевых эффектов PPARg.
- Тем не менее, данные генетические и фармакологические эксперименты обнаруживают, что Gpr132 является весьма важной деталью паззла и необходимым медиатором эффектов PPARg в макрофаге, поскольку отсутствие Gpr132 отменяет влияние PPARg или росиглитазона на опухоль.
- Gpr132 становится не только ключевым регулятором PPARg, но и новой мишенью в терапии рака.
Работы с люциферазой позволяют сделать заключение о том, что:
- PPARg напрямую ингибирует транскрипционную активность промотора Gpr132 (исследование ChIP также демонстрирует, что PPARg связывается с промотором Gpr132, что приводит к снижению уровня H3K9Ac – активного транскрипционного гистона на стартовом сайте транскрипции Gpr132).
- Тем не менее, вспомогательные механизмы, такие как изменения стабильности мРНК, могут также влиять на снижение уровня мРНК в Gpr132 под воздействием PPARg.
Опухолевые клетки вступают в тесные взаимодействия с ОАМ для дальнейшей пролиферации и выживания. В частности, воздействие на инфильтрирующие макрофаги для регулирования их количества и свойств, может значительно снизить степень злокачественности опухоли. Данное исследование демонстрирует, что этот эффект может быть достигнут как активацией PPARg, так и методом ингибирования Gpr132.
- Путем изучения механизмов, которые используют макрофаги в процессе развития воспаления и опухоли, могут быть найдены новые диагностические инструменты, а также разработаны инновационные противоопухолевые и противовоспалительные терапевтические средства.
- К примеру, по уровню экспрессии PPARg и Gpr132 в макрофаге можно предполагать не только степень агрессивности опухоли, но также ее фармакологический ответ на терапию росиглитазоном или ингибиторами Gpr132.
- Полученные данные объясняют, почему росиглитазон обладает противоопухолевой активностью лишь при определенных типах рака: резистентными к терапии остаются онкологические заболевания, при которых число макрофагов остается относительно низким или присутствуют PPARg-негативные макрофаги.
- Позитивная ассоциация Gpr132 с процессами воспаления и РМЖ у человека, а также угнетение экспрессии Gpr132 при применении росиглитазона и связанные с этим антиопухолевые эффекты характеризуют Gpr132 как потенциально эффективный терапевтический инструмент (рис. 5O–P).
- Определение уровня экспрессии Gpr132 можно использовать также в качестве онкомаркера наряду с уже известными маркерами (21 ген), представленнымы в диагностическом тесте Oncotype DX (рис. 4B).
Суммируя все вышесказанное:
-
PPARg в макрофаге – важнейший медиатор противоопухолевых эффектов росиглитазона.
-
Gpr132 — ген-мишень, контролирующий функции PPARg.
-
Gpr132 – провоспалительный и проопухолевый фактор макрофага и, соответственно, терапевтическая мишень.
Благодаря этим новым данным, значительно улучшилось понимание макрофагальной регуляции, биологии опухолевого микроокружения, в том числе PPARg и Gpr132, что крайне важно для терапии рака, диабета и заболеваний воспалительного генеза.
Материалы и методы
Мыши
Были описаны мыши линии C57BL/6J с флоксированным геном PPARg. Те же линии мышей с нокаутом по гену Gpr132 были взяты из лаборатории the Jackson Laboratory. Все мыши получали одинаковое стандартное питание и содержались в цикле: 12 часов с освещением и 12 часов в темноте.
- Мыши с флоксированным геном PPARg скрестили с мышами линий Tie2-Cre и Lysozyme-Cre (трансгенных по Cre-рекомбиназе) для выведения особей mf-g-KO (с нокаутом по PPARg в макрофагах).
- Затем мышей Tie2-g-KO скрестили с Gpr132-KO и получили мышей с нокаутом по обоим генам: mf-g/Gpr132 double KO.
- Результаты в группе Tie2-g-KO оказались менее специфичными, чем у Lyz-g-KO.
- Все эксперименты проводились на одонопомётных мышах.
- Оценка результатов была проведена при помощи SAS 9.3 на платформе TS X64_7PRO.
- Эксперименты на мышах проводились в соответствии с протоколами, одобренными Комитетом по биоэтике (IACUC).
Культуры опухолевых клеток и макрофагов
- Для макрофагов селезенки и костного мозга, спленоциты и костный мозг мышей культивировали на бессывороточной среде Игла, модифицированной по способу Дульбекко (DMEM).
- После пассирования через 40 µm клеточный фильтр, клетки культивировались в течение 6 дней на среде DMEM + 10% FBS (фетальная бычья сыворотка) + 20 ng/ml M-CSF (колониестимулирующий фактор макрофага) для дифференцировки макрофагов.
- При помощи лентивирусной трансдукции, удалось добиться гиперэкспресии Gpr132.
- Все линии клеток были аутентифицированны при помощи короткого концевого повтора (STR), а также было установлено отсутствие микоплазмы.
- В совместных культурах клеток (макрофаги+опухолевые клетки), клетки костного мозга и селезенки мышей предварительно культивировались в течение 9 дней при помощи 20 ng/ml M-CSF для дифференцировки в макрофаги. К ним добавили клетки линии 4T1.2, меченные люциферазой.
- На завершающем этапе, клеточный лизат был забран для люциферазного анализа, чтобы оценить рост опухолевых клеток.
- За 24 часа до добавления в культуру опухолевых клеток, макрофаги были обработаны росиглитазоном (1 µm).
Ортотопическая инъекция клеток РМЖ
- Клетки линии EO771 (2.5х105 или 5х105) были введены в жировую ткань молочной железы самкам мышей в возрасте 6-8 недель.
- Каждые 2-3 дня производились измерения длины и ширины опухоли, вычислялся ее объем по формуле V =(Длина * ширина* ширина )/2.
- Линия клеток Py230 была получена из спорадических опухолей самок трансгенных мышей линии C57BL/6 MMTV-PyMT.
Иммунофлуоресцентное окрашивание
- Ткани опухоли были извлечены от мыши через 3 недели после инъекции опухолевых клеток. Прежде, чем обработать клетки опухолей антителами, опухоли были заморожены. Приготовленные срезы зафиксировали ацетоном.
- Срезы опухолей, стабилизированные 2% раствором BSA (бычий сывороточный альбумин), были выдержаны с флуоресцирующими (FITC) анти- CD11b или анти- F4/80 антителами, крысиными моноклональными анти-эндомуциновыми антителами, крысиными поликлональными анти-фосфо-гистон H3 или моноклональными анти- Ki67 клетками кролика.
- После отмывания с забуференным фосфатом физраствором (PBS), срезы были обработаны антителами IgG-FITC козьи к мышиным или козьи к кроличьим.
- После последующего отмывания с PBS, срезы были закреплены в среде (Vectashield), содержащей флуоресцентный краситель DAPI.
Анализ экспрессии генов
- Образцы тканей были заморожены при помощи жидкого азота и хранились при температуре -80 °C.
- Экстракция РНК была произведена при помощи раствора Тризола в соответствии с протоколом производителя.
- Чтобы удалить всю геномную ДНК, РНК была обработана дезоксирибонуклеазой без РНК-аз, а затем транскрибирована в комплементарную ДНК при помощи комплементарной ДНК высокой производительности ABI RT Kit (Invitrogen).
- кДНК была проанализирована при помощи количественной ПЦР в режиме реального времени с использованием приборов для определения последовательностей (Applied Biosystems 7700).
- Каждая реакция была выполнена трижды в формате 384.
- Экспрессия генов мышей была нормализована мышью L19.
- Экспрессия генов человека – человеческой глицеральдегид–3–фосфатдегидрогеназой (GAPDH).
- Анти- Gpr132 антитела были проверены при помощи клеток линии Gpr132-KO и использованы для обнаружения Gpr132 методом иммуноблота.
Иммуногистохимия
- Тканевые матрицы были приобретены в US Biomax, Inc. и содержали нормальные и злокачественные клетки молочной железы человека.
- Иммуногистохимическое исследование (ИГХ) выполнялось согласно вышеописанным этапам:
- депарафинизация ксиленом;
- дегидратация этанолом;
- инкубация в буфере с восстановленным антигеном в течение 1 часа при температуре 95 °C;
- обработка 3% раствором перекиси водорода в течение 10 минут;
- образцы были обработаны 5% обезжиренным молоком в течение 1 часа при комнатной температуре, затем соединены с анти- Gpr132 антителами при 4°C на 1 ночь, а затем на 30 минут при комнатной температуре со вторичными конъюгированными с HRP антителами (1:200).
- Иммунное окрашивание было выполнено при помощи диаминобензидина.
- Уровень экспрессии Gpr132 был определен слепым методом полуколичественно соответственно интенсивности и распределению окрашивания при помощи шкалы иммунореактивов:
- ИГХ= интенсивность окрашивания (отрицательно-0; слабо=1; умеренно=2; интенсивно=3)*процент позитивных клеток (0%=0; 0-25%=1; 25–50% = 3; 75–100% = 4).
Анализ данных TCGA (геномный атлас рака)
Клинические данные секвенирования РНК инвазивной карциномы молочной железы были получены из TCGA и протестированы на совпадения. Экспрессия генов GPR132, CCL2 (MCP-1), MMP9 и PTGS2 (COX-2) анализировалась методом линейной регрессии.
Статистический анализ
Все статистические исследования были выполнены с t-тестом и представлены как среднее ± стандартное отклонение (SD), если не отмечено иначе. Для экспериментов in vivo с ≥ 3 группами, статистические исследования были выполнены при помощи дисперсионного анализа (ANOVA) и сравнительного анализа пар. Значения p определялись как статистически значимые при *p<0.05; **p<0.01; ***p<0.005; ****p<0.001; не значимые p>0.05.
Источник:
- Cheng WY, Huynh H, Chen P, Peña-Llopis S, Wan Y. Macrophage PPARγ inhibits Gpr132 to mediate the anti-tumor effects of rosiglitazone. Tontonoz P, ed. eLife. 2016;5:e18501. doi:10.7554/eLife.18501.